On May 1st 2020, a new finding entitled “In vivo stress granule misprocessing evidenced in a FUS knock-in ALS mouse model” was published in current issue of Brain by Dr. Yichang Jia’s group in the School of Medicine at Tsinghua University, IDG/McGovern Institute for Brain Research at Tsinghua, and Tsinghua-Peking Joint Center for Life Sciences. In this work, Xue Zhang et al. described a new and rational mouse model for ALS and provided new insights into the disease mechanisms.
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a devastating neurodegenerative disease that affects at least 16,000 at any given time in USA (http://www.alsa.org/) and estimated 65,000-100,000 in China. It leads to motor neuron nerve loss in brain and spinal cord that control your muscles. Therefore, as the disease progresses, it gets harder for patients to walk, talk, eat, and breathe. Many people live with the disease for only 2-5 years from the time of their diagnosis and currently there is no cure for the disease, partially due to our poor understanding of the disease mechanisms.
In 2006, Dr. Virginia Lee at UPenn first disclosed TDP-43 pathology in ALS and Frontotemporal dementia (FTD) (Neumann et al., 2006). Subsequent studies revealed that the mutations of TDP-43 and FUS, two structurally similar RNA binding proteins (RBPs), are associated with familiar and sporadic ALS (Cruz and Cleveland, 2011). Strikingly, the TDP-43 and FUS pathologies, majorly mislocalized and ubiquitin-positive these two RBPs (Dormann and Haass, 2011), are not only found in ASL and FTD as Dr. Virginia Lee disclosed but also in a large proportion of other neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases, underscoring the dysfunction of these RBPs are common in the etiology of the diseases. However, how these RBP dysfunction leads to the diseases is still largely unknown.
After identification of mutations of TDP-43 and FUS in ALS, scientists modeled the disease by using mouse and transgenic approach, which ectopically overexpressed the ALS mutant proteins many times in mouse. In fact, ALS patients carry a point mutation rather than overexpression of these RBPs. Therefore, Xue Zhang et al. generated a new mouse ALS model which carries an ALS point mutation to reflect the disease nature. However, this new model does not display paralysis and shorter lifespan as seen in the patients, suggesting the second hit required for the pathogenesis.
Emerging evidence reveals that TDP-43 and FUS among other RBPs with low complexity domain are involved in a cellular process called stress granule (SG), a RNA/protein complex assembled when cell faces stress to cease the cytoplasmic mRNA translation but disassembled when the stress is withdrawn. These previous findings supposed that ALS mutant TDP-43 and FUS affect the processing of SG in the disease pathogenesis (Aulas and Velde, 2015). However, whether and how reasonable level of ALS mutant RBP affects SG dynamics in diseased neurons are largely unknown. The new model generated by Xue Zhang et al. allows scientists to answer the above question. Using the new model, Xue Zhang et al. documented that the mutant but not wildtype FUS moves into SG and majorly affects mutant SG disassembly when motor neurons facing oxidative stress challenge (Figure 1). More importantly, they challenged the mutant mouse with an oxidative stress inducer arsenite and observed that the challenge led to ALS-like pathologies and severe motor defects in the mutant but not wildtype animals. Xue’s findings for the first time provide experimental support that environmental insult and gene mutation---two hits---synergistically contribute to ALS pathogenesis.
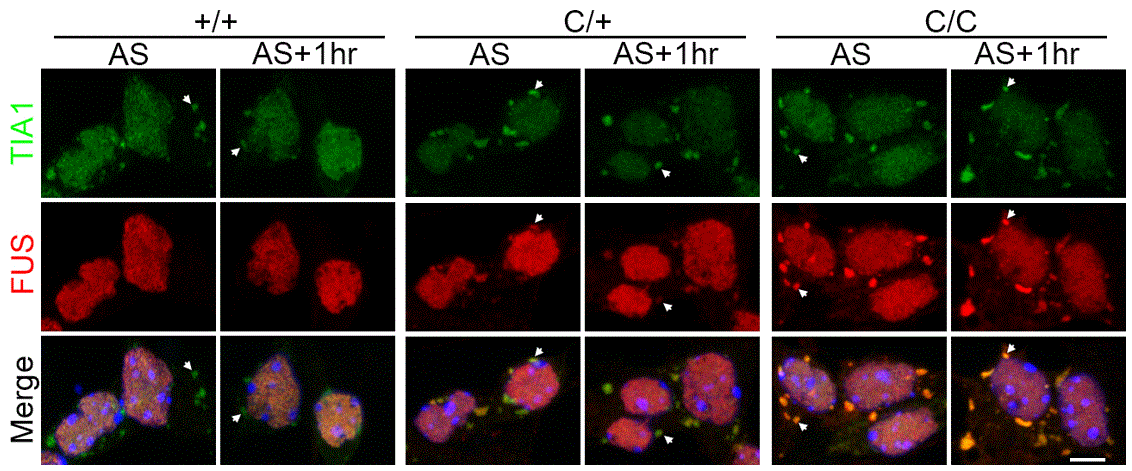
Figure1. ALS mutant FUS (R521C) moves into TIA1-positive stress granules in cultured motor neuron. C/+, heterozygous for the R521C mutation; C/C, homozygous for the mutation. AS, treatment of arsenite, an oxidative stress inducer; AS+1hr, withdraw of AS for 1 hour. Arrow head, stress granules.
Although SG misprocessing is hypothesized to play a role in ALS etiology, no evidence supports that it occurs in the diseased brain and is pathogenic. To address the questions, Xue Zhang et al. faced a series of challenges, including 1) how to induce stress insult in brain; 2) how to label, trace, and monitor the SG in brain; 3) how to analyze and reconstruct the in vivo SG signals, because nobody had done in vivo SG imaging before. After overcoming all above obstacles, Xue Zhang et al. for the first time visualized and traced the SG processing in the same neurons before and after stress challenge in a living mouse brain for weeks and observed that SG misprocessing in the mutant motor cortex but much less in that of wildtype animals. In addition, they documented that the neuron with more severe SG processing defect is prone to loss in the diseased brain (Video 1). The in vivo evidence provided by Xue Zhang et al. for the first time demonstrated that SG misprocessing is pathogenic. Xue’s prototype research provides the field a rational ALS model and a brand-new paradigm to trace the SG processing in a living animal brain, which will help scientists on the translational research and mechanistic studies for ALS.
Video 1. In vivo evidence for disappearance of mutant (C/C) neurons that carry severe abnormalities in TIA1-positive granule processing. The signals shown are TIA1-EGFP, which was employed to visualize and trace the in vivo stress granule before and after stress treatment. Pretreatment, before stress challenge; hrs, hours; ws, weeks. Note a mutant neuron disappeared by the end of our experiment (3ws).
Ph. D. student Xue Zhang is the first author of this work. Drs. Fengchao Wang at NIBS (National Institute of Biological Sciences, Beijing) and Jisong Guan at ShanghaiTech University contribute to this work in generation of mouse model and two-photon imaging. This work is supported by School of Medicine at Tsinghua University, Peking-Tsinghua Joint Center for Life Sciences, IDG/McGovern Institute for Brain Research at Tsinghua, the National Natural Science Foundation of China (NSFC), and Amyotrophic Lateral Sclerosis Association (ALSA).
References:
Aulas, A., and Velde, C.V. (2015). Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Frontiers in Cellular Neuroscience 9, 423-423.
Cruz, S.D., and Cleveland, D.W. (2011). Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current Opinion in Neurobiology 21, 904-919.
Dormann, D., and Haass, C. (2011). TDP-43 and FUS: a nuclear affair. Trends in neurosciences 34, 339-348.
Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., and Clark, C.M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130-133.
Paper link:https://academic.oup.com/brain/advance-article-abstract/doi/10.1093/brain/awaa076/5827862?redirectedFrom=fulltext