Highlights
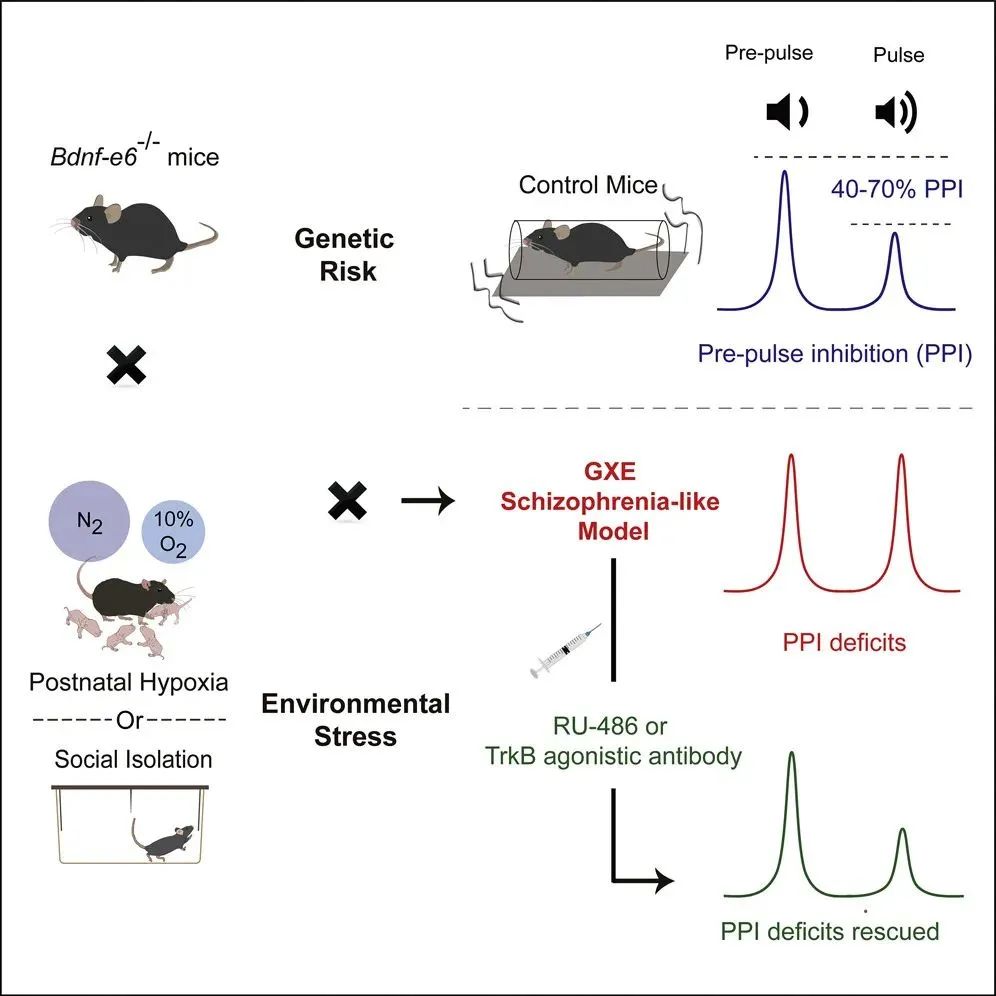
•Bdnf-e6−/− combining early-life stress results in schizophrenia-like phenotypes
•Neither early-life stress nor Bdnf-e6 deficiency alone causes these abnormalities
•Corticosterone treatment to Bdnf-e6−/− mice also induces PPI deficits
•PPI deficits can be rescued by treatment with RU-486 or TrkB agonistic antibody
While schizophrenia pathogenesis involves both genetic and environmental factors, their specific combinations remain ill-defined. Here we show that deficiency in promoter VI-driven BDNF expression, combined with early-life adversity, results in schizophrenia-like endo-phenotypes. Promoter VI mutant mice (Bdnf-e6−/−), when exposed to postnatal stress including hypoxia or social isolation, exhibited deficits in social interactions, spatial memory, and sensorimotor gating reflected by prepulse inhibition (PPI). Neither early-life stress nor Bdnf-e6 deficiency alone caused these abnormalities. Moreover, postnatal stress increased blood corticosterone levels of wild-type mice, and administration of corticosterone to Bdnf-e6−/− mice without early-life stress also resulted in PPI deficits and social dysfunction. Finally, the PPI deficits in postnatally stressed Bdnf-e6−/− mice were rescued by treatment with the corticosterone antagonist RU-486, or the BDNF mimetic TrkB agonistic antibody. Thus, we have identified a pair of genetic and environmental factors contributing to schizophrenia pathogenesis and providing a potential strategy for therapeutic interventions for schizophrenia.
|
Paper Link:https://www.sciencedirect.com/science/article/pii/S2589004222008811?via%3Dihub
|