Abstract
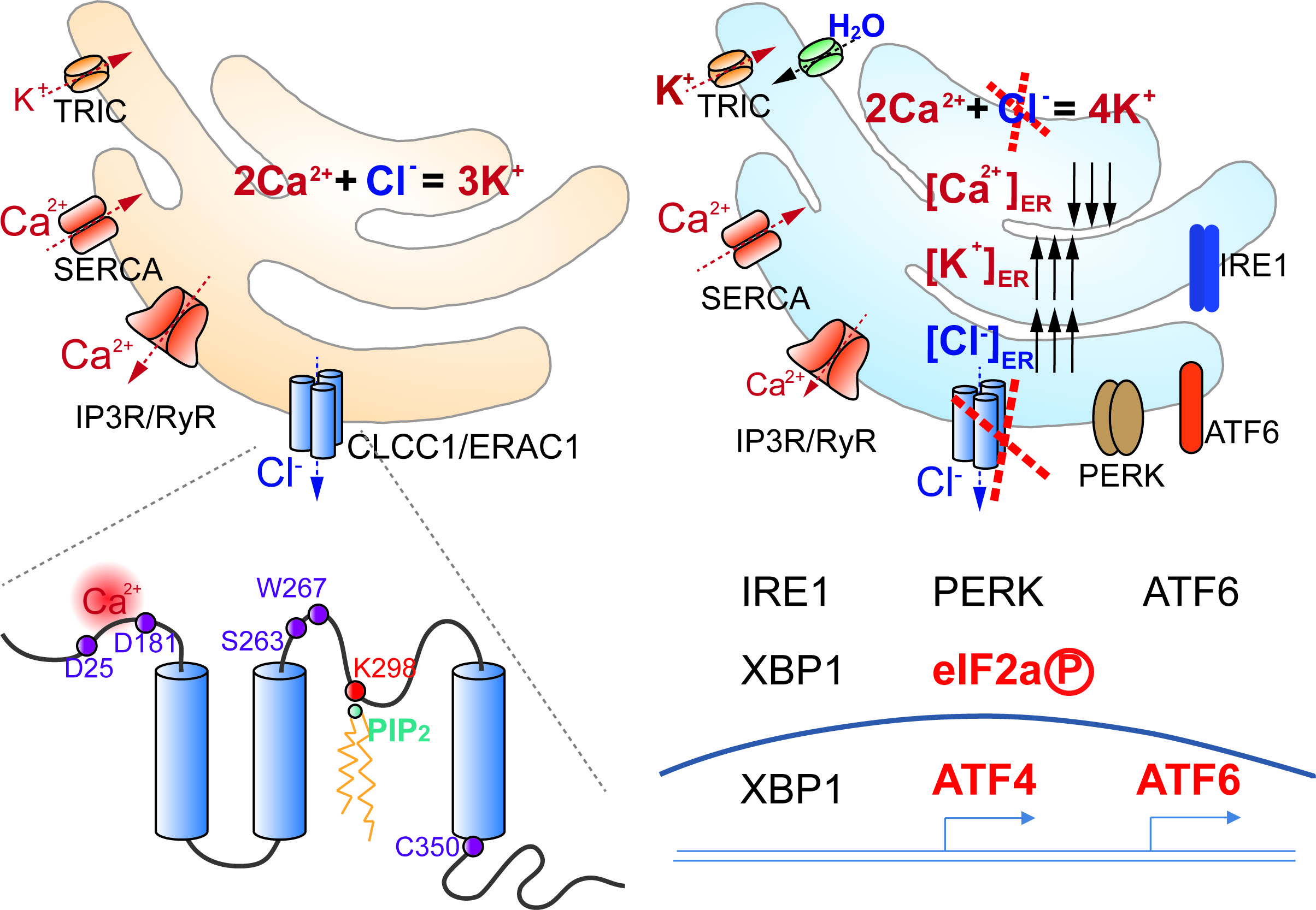
Although anion channel activities have been demonstrated in sarcoplasmic reticulum/endoplasmic reticulum (SR/ER), their molecular identities and functions remain unclear. Here, we link rare variants of Chloride Channel CLIC Like 1 (CLCC1) to amyotrophic lateral sclerosis (ALS)-like pathologies. We demonstrate that CLCC1 is a pore-forming component of an ER anion channel and that ALS-associated mutations impair channel conductance. CLCC1 forms homomultimers and its channel activity is inhibited by luminal Ca2+ but facilitated by phosphatidylinositol 4,5-bisphosphate (PIP2). We identified conserved residues D25 and D181 in CLCC1 N-terminus responsible for Ca2+ binding and luminal Ca2+-mediated inhibition on channel open probability and K298 in CLCC1 intraluminal loop as the critical PIP2-sensing residue. CLCC1 maintains steady-state [Cl–]ER and [K+]ER and ER morphology and regulates ER Ca2+ homeostasis, including internal Ca2+ release and steady-state [Ca2+]ER. ALS-associated mutant forms of CLCC1 increase steady-state [Cl–]ER and impair ER Ca2+ homeostasis, and animals with the ALS-associated mutations are sensitized to stress challenge-induced protein misfolding. Phenotypic comparisons of multiple Clcc1 loss-of-function alleles, including ALS-associated mutations, reveal a CLCC1 dosage dependence in the severity of disease phenotypes in vivo. Similar to CLCC1 rare variations dominant in ALS, 10% of K298A heterozygous mice developed ALS-like symptoms, pointing to a mechanism of channelopathy dominant-negatively induced by a loss-of-function mutation. Conditional knockout of Clcc1 cell-autonomously causes motor neuron loss and ER stress, misfolded protein accumulation, and characteristic ALS pathologies in the spinal cord. Thus, our findings support that disruption of ER ion homeostasis maintained by CLCC1 contributes to ALS-like pathologies.
|
Paper Link: https://doi.org/10.1038/s41422-023-00798-z
|